This article was originally published by STAT on October 1, 2018.

During my 19 years as a research scientist at the Upjohn Company, the regulatory burden imposed by the open-ended Drug Amendments Act of 1962 was mushrooming. My coworkers and I joked that because of this dramatic change to the mission of the FDA, made largely as a response to the European thalidomide crisis, we spent so much time meeting the FDA’s development demands that we had little time to discover new drugs.
Like most gallows humor, such comments masked the very real concern that instead of finding drugs that would save lives, we were checking regulatory boxes. We never dared to hint at these concerns in public for fear that the FDA might retaliate by dragging its feet to delay our company’s drug approvals.
The amendments gave the FDA the authority to require proof of effectiveness — not just safety — before approving a new drug. It could dictate which manufacturing standards needed to be met before animal testing, which animal tests needed to be done before clinical trials, and which human studies were needed for approval. The amendments also gave the FDA jurisdiction over advertising and labeling.
Before 1962, a drug company submitted its data on a new drug to the FDA and could market the drug after six months if the agency didn’t object. The amendments required a “sign off” by an individual regulator, putting that person’s neck in the proverbial noose. Whether the amendments have improved the drug-development process and protected the public’s health continues to be questioned.
In a series of studies stretching from 1979 to the present, Joseph A. DiMasi, Henry G. Grabowski, and Ronald W. Hansen confirmed what my co-workers and I already knew: the cost of taking a new drug, often referred to as a new molecular entity, from the lab bench to the market has soared in the last several decades, primarily due to increasing regulatory demands, as shown in the graph below. These costs must be passed on to consumers as higher prices at the pharmacy if drug companies are to remain solvent.
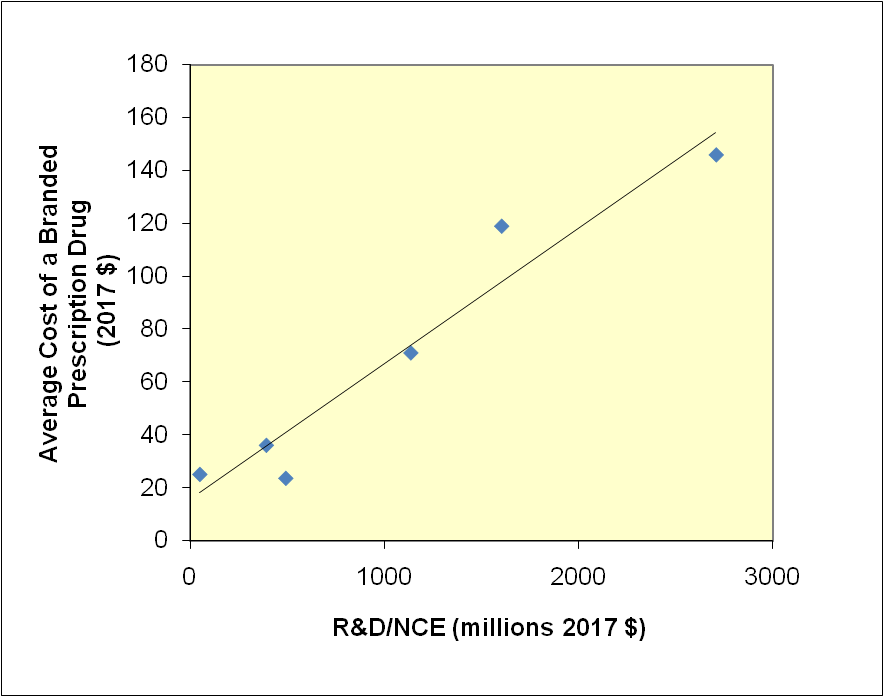
Did the additional work required by the FDA give us more safety? The evidence is hardly compelling. Using data from several published sources (references available upon request), I calculated that the rate at which FDA-approved drugs were withdrawn from the market because of unacceptable side effects averaged 2.5 percent from 1947 to 1961 and 3.3 percent from 1962 to 2011. Indeed, properly prescribed drugs may now be the fifth largest cause of death in the United States.
Are drugs more effective now than they were before 1962? Researchers have estimated that the 1962 amendments might have saved consumers at most 10 percent on their pharmacy bills by keeping ineffective drugs off the market, while increasing drug costs in the neighborhood of sevenfold to eightfold. In other words, ineffective drugs didn’t survive in the pre-amendment marketplace even without the regulatory requirement that effectiveness be documented by placebo-controlled studies.
In a 1975 analysis, University of Chicago economist Lester G. Telser showed that drug prices adjusted for inflation decreased 32 percent from 1949 to 1961. Yet in the decades after the 1962 amendments, drug prices increased as development costs rose in response to the ever-growing regulatory demands.
Between 1948 and 1961, pharmaceutical companies took an average of about four years to take a new drug to market. By the 1980s, the development time had increased to 14 years and still averages 10 to 15 years.
Some people can’t wait that long for new treatments. In the late 1980s and early 1990s, AIDS patients hired chemists to make the same drugs the pharmaceutical industry was working on. By the time the FDA gave companies permission to start clinical trials, many AIDS patients in the United States who wanted these drugs had already gotten them via the black market and developed resistance to them, similar to what we see now with antibiotics.
Desperate patients are still taking matters into their own hands rather than die waiting as new drugs creep through the lengthy development process. Today, patients with amyotrophic lateral sclerosis are making in their kitchens drugs that look promising, and do-it-yourself kits are being developed for home manufacture of drugs for asthma, hepatitis C, and other diseases.
Innovation is lost as funds are shifted from the discovery of new drugs to the development of those in the pipeline. About half of new drugs that are abandoned in mid- to late-stage development are lost for economic reasons because the companies feel they won’t be profitable enough to justify taking the drug to market — sometimes even for drugs that hold clinical promise. Manufacturers are rapidly abandoning essential research in antibiotics, for example, as they realize their short treatment window makes cost recovery, let alone profits, unlikely.
Many more new drugs are abandoned even before clinical studies begin. The FDA once contacted me to support the development of prostaglandins for liver disease after seeing my patent filing for that indication. For truly novel approaches, like prostaglandins for liver disease, the unknowns are great enough that a mistake in dose, duration of treatment, treatment schedule, number of patients, and the like can mean that effectiveness studies might not have the statistical significance the FDA demands. If these studies, which last for years, have to be repeated, the patent may expire and cost recovery becomes virtually impossible. Even with the enthusiastic support of the FDA, Upjohn management decided it was too risky to even attempt development of prostaglandins for liver disease.
The drug development system that emerged from the 1962 amendments, which were intended to increase drug safety and effectiveness, have given us minimal benefit at great cost, not just in money but in lives. Using estimates of how many lives are saved by the drugs on the market today, I calculate in my latest book, “Death by Regulation,” at least a five-year loss in life expectancy, primarily due to longer development times after 1962 and loss of about 50 percent of our innovations, assuming that they save only 25 percent as many lives as today’s drugs do. Excess regulation has side effects that are just as deadly as bad drugs. All of us — regulators, politicians, drug company employees, patients, and doctors — are affected.
Reversing the 1962 amendments could slash new drug costs virtually overnight, stimulate innovation, and add years to our lives. The amendments demand that drugs be safe and effective, but apparently don’t meet these same standards themselves. Perhaps deadly regulations should be withdrawn, just as unsafe drugs are.
Mary J. Ruwart, Ph.D., is a former research scientist with The Upjohn Company and currently chairs a for-profit institutional review board. She consults for nutraceutical firms, biotech startups, and occasionally testifies as an expert witness. She is also the author of “Death by Regulation: How We Were Robbed of a Golden Age of Health and How We Can Reclaim It” (SunStar Press, 2018).








While this is all true, I have come to learn that pharmaceutical drugs mostly make you feel better temporarily but harm you in the long run. Same with most surgeries. I recently had a skin cancer. I had no idea it was a skin cancer but it was ugly and I wanted to get it removed. It took two months to get someone to look at it, another month to get a biopsy, another month to get the results and another month to get a surgery appointment. Well, before I knew it was a cancer, I read about a protocol using food grade hydrogen peroxide that cures many ailments. Since I have chronic Lyme disease, Lupus, and have symptoms of ALS, I decided to give it a shot. I had nothing to lose. In a couple of weeks, I noticed my lesion was going away, so I kept doing the protocol. I thought if I could get this ugly thing to go away with hydrogen peroxide with no surgery, that would be wonderful. I didn’t think insurance would pay for it since I didn’t know it was cancer. It took 2 1/2 months for it to go away completely so I was able to make it go away on its own without surgery before a surgeon could schedule and do a surgical procedure to remove it!
I have more great stories like this. I have learned the best way to stay healthy and well is to stop going to see your doctor, change your diet, get off all drugs if possible. I am on a new protocol now using something different and I am seeing my symptoms of ALS disappear. I decided I didn’t need a diagnosis since doctors have no cure for ALS and this is cheap to try, under $100. I think now that most of the medical system is a total sham. I used to believe in them and trust them but no more. I lost my gall bladder, my thyroid, had an unnecessary hysterectomy, and a fundoplication that nearly killed me, all of which were totally reversable with natural treatments and dietary changes. No one ever told me that.
Sorry to be so late in replying. It looks like I didn’t get a notification of your e-mail.
I agree with you; prevention or natural cures are best. That said, at some point, they will fail. If they didn’t, we’d all be able to live forever. The reason your doctors don’t know about them is that the FDA will prosecute any company that claims therapeutic effects for a natural product, including food, unless the manufacturer goes through the same 10-14 year process that new drugs do.
Most drugs are chemical cousins of natural products. Because they are chemically designed to last longer in your body than the natural substances, they can extend life when prevention fails. By the same token, these powerful substances will have side effects in some people and will limit their usefulness.
Like you, I focus on natural alternatives but will take drugs if I find them useful. So far, so good. 🙂
I blog frequently and I truly thank you for your content. This great article has truly peaked my interest.
I’m going to take a note of your site and keep checking for new details
about once per week. I opted in for your Feed as well.